Genomics Learning Center
Learn about next generation technologies that enable researchers to obtain unprecedented insights into complex biological systems, whether cells in suspension or in tissue.

Applications
The acquisition of multiomic data is becoming increasingly important in single-cell RNA sequencing (scRNAseq) and spatial transcriptomics (ST) studies. Multiomic approaches offer a more holistic view of cellular identities and functions. Technological advancements such as Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) and RNA Expression and Protein Sequencing (REAP-seq), have empowered researchers to simultaneously measure the expression of hundreds of cell surface proteins during scRNAseq experiments. Here are several reasons why the addition of protein expression measurements during scRNAseq and ST studies is beneficial:
Enhanced cellular characterization
Proteins are the functional molecules that carry out various biological processes within cells. By measuring protein expression in addition to mRNA expression, researchers can gain a more comprehensive understanding of cellular states and functions.

Validation of RNA expression
scRNAseq provides information about gene expression, but it doesn’t directly measure protein abundance. By simultaneously measuring protein expression, researchers can explore the correlation between RNA and protein expression levels, providing a more accurate assessment of cellular states.
Biomarker discovery
scRNAseq combined with protein measurements can facilitate the identification of specific protein markers associated with different cell types, disease states, or therapeutic responses. These protein markers can have diagnostic, prognostic, or therapeutic implications.
What is scRNAseq?
Single-cell RNA-seq (scRNAseq) or Single-cell Transcriptomics is a powerful technique that allows researchers to analyze gene expression at single-cell resolution. scRNAseq provides transcriptome profiling of up to millions of cells per experiment. scRNAseq offers valuable insights into cellular identity, behavior, disease mechanisms, and biomarker identification, making it a powerful tool for researchers in diverse fields. The benefits of scRNAseq include:
Unveiling cellular heterogeneity
scRNAseq enables the identification and characterization of distinct cell types, subpopulations, and rare cell states within complex cell mixtures.
Revealing transcriptional dynamics
scRNAseq can aid in uncovering gene regulatory networks, cell fate determination, cell differentiation, and understanding cellular responses to external stimuli.
Biomarker discovery and precision medicine
scRNAseq has the potential to identify novel biomarkers specific to different cell types or disease states.
Drug discovery and development
scRNAseq offers opportunities for a deeper understanding of drug responses and evaluating the efficacy and safety of potential therapeutics.
scRNAseq workflow
- Cell suspension is generated.
- Using one of a variety of methods, cells are partitioned into single cell compartments.

- mRNA and other cellular contents are made accessible to downstream molecular biology via cell lysis or cell permeabilization.
- Released mRNA undergoes reverse transcription, during which, each transcript is tagged with a cell ID DNA barcode. All transcripts from an individual cell are tagged with the same cell ID DNA barcode, which is unique from cell to cell.

- DNA and appended cell barcodes are pooled and prepared into double stranded DNA sequencing libraries.

- Next Gen DNA Sequencing is used to read out transcript and cell barcode information from each cell.

- Gene expression data is used to provide phenotypes and functional states for individual cells.
scRNAseq techniques
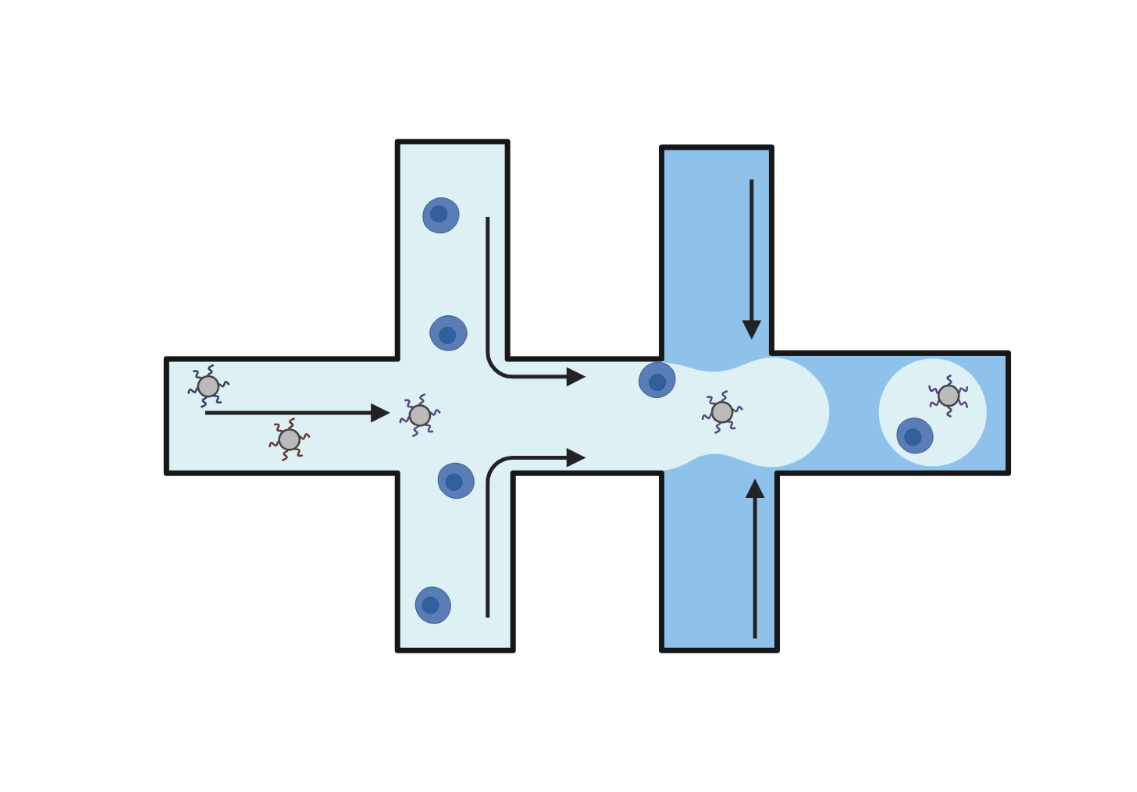
Droplet
Droplet-based technologies use microfluidics to encapsulate individual cells and barcoded beads into aqueous droplets suspended in oil. Within each droplet cells are lysed, and reverse transcription takes place, resulting in barcoded cDNA libraries. These libraries are then pooled and sequenced. Droplet-based methods offer high-throughput capabilities, enabling the analysis of up to a million cells in a single experiment.
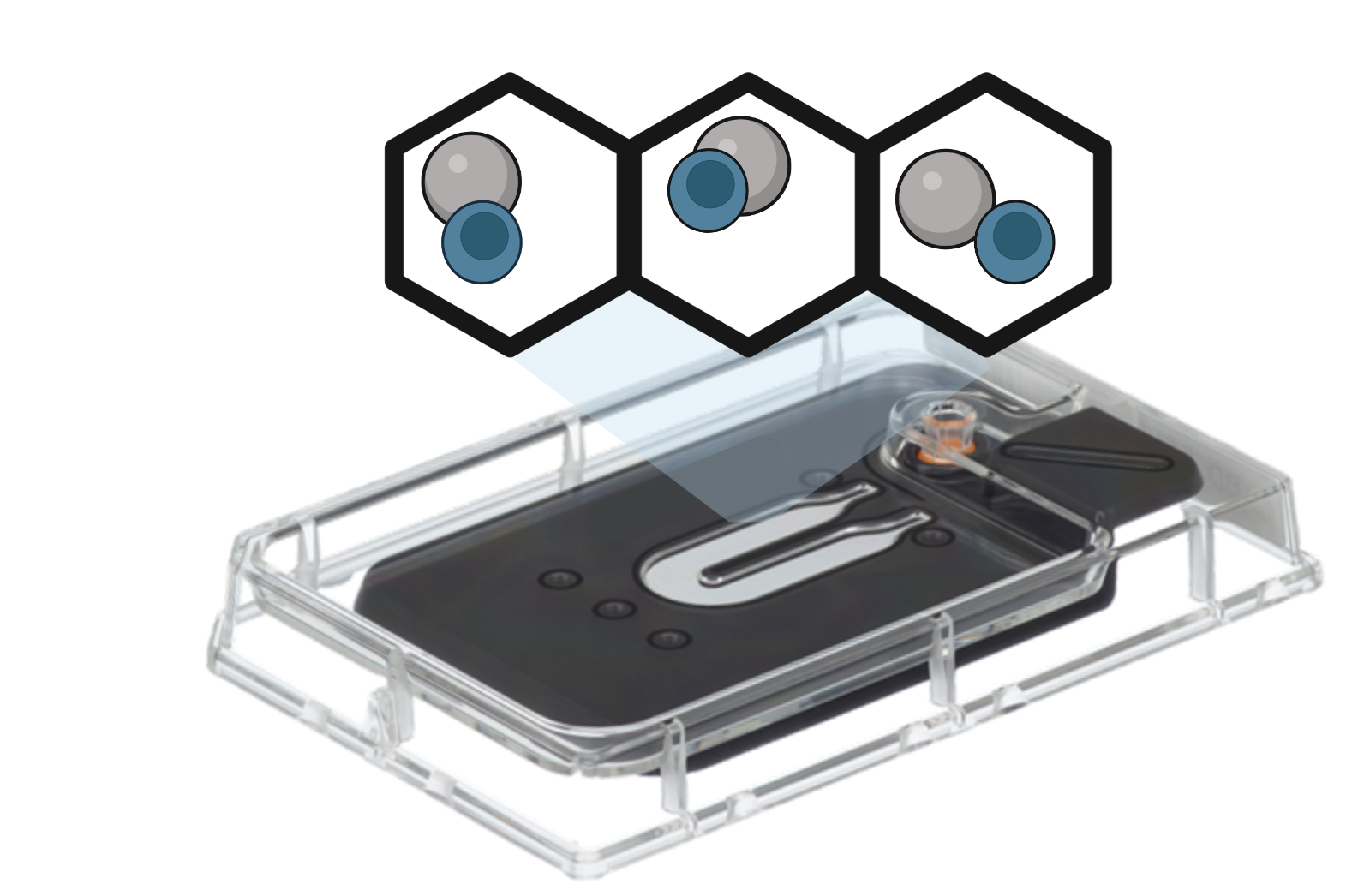
Microwell
Microwell-based technologies utilize microfluidic devices and specialized plates that contain arrays of cell capture wells. Individual cells are isolated into separate microwells along with mRNA capture beads. Cell lysis, reverse transcription, and barcoding occur within each well. The resulting barcoded cDNA libraries are subsequently pooled and sequenced.
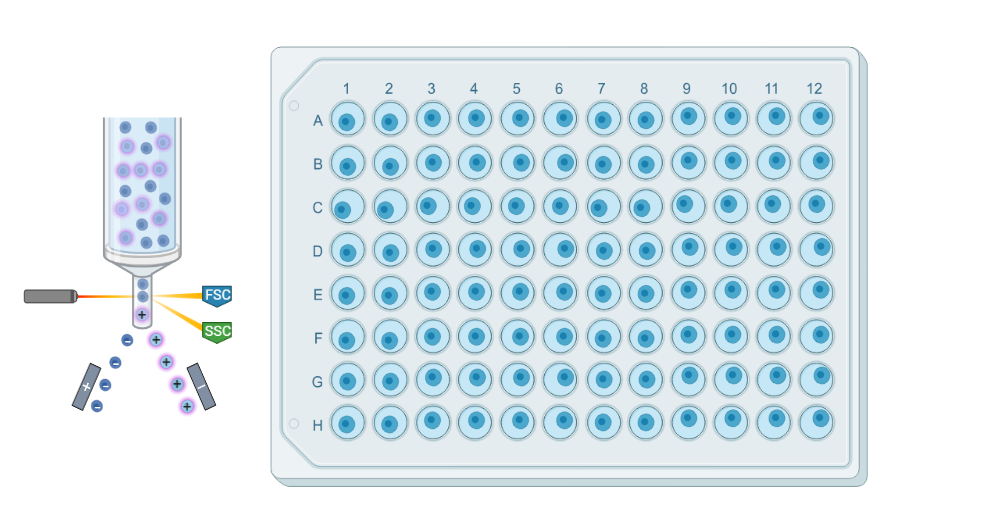
Plates
Plate-based scRNAseq methods involve isolating via cell sorting and processing individual cells in a traditional plate format. Cells are typically captured in 96- or 384-well plates, and reverse transcription and library preparation are performed within each well.
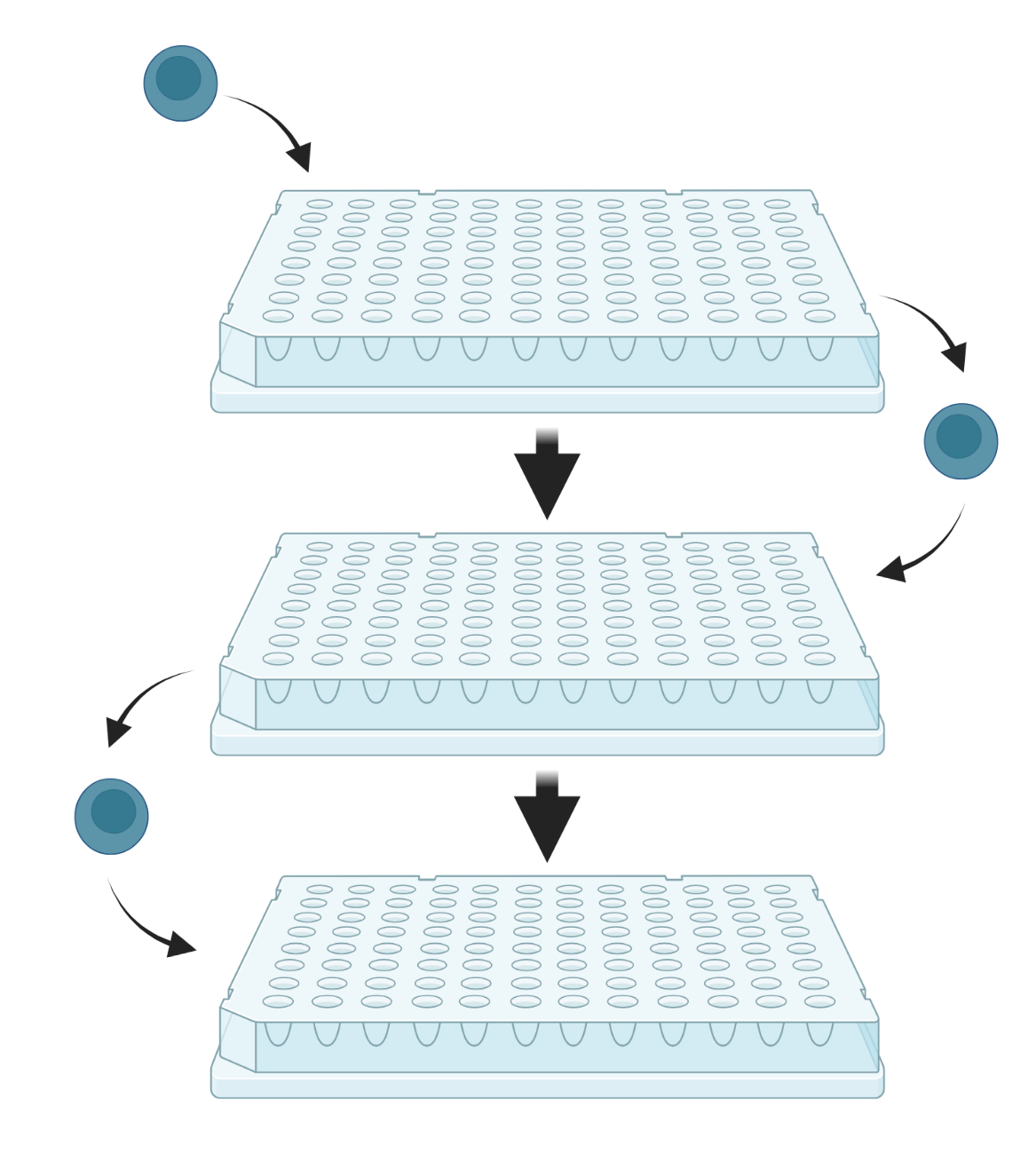
Split-pool
Split-pool-based methods rely on the combinatorial barcoding of individual transcriptomes through a series of splitting and pooling events across several multiwell plates. The plates contain unique oligonucleotide barcodes in each well, enabling sequential barcoding as cells move from well to well. The resulting barcoded cDNA is subsequently pooled and sequenced.
What is Spatial Transcriptomics?
Spatial Transcriptomics (ST) represents a group of powerful methodologies for the visualization and quantification of gene expression while preserving the spatial context of transcripts. Transcriptomics techniques, such as scRNAseq, provide a wealth of information about gene expression, but they lose the spatial context of where in the tissue these genes are being expressed. ST methodologies seek to address this limitation by enabling researchers to visualize mRNA molecules directly in situ or by capturing mRNA in a manner that preserves the physical coordinates of the mRNA. ST is becoming increasingly popular for several reasons:
Preservation of Spatial Context
ST provides a comprehensive view of gene expression while maintaining spatial context. This is crucial because cells within tissue exist in a complex environment where they’re influenced by contact with other cells and environmental factors.
Understanding Complex Tissues
ST allows researchers to study complex tissues and organs, such as the brain and tumors, by enabling them to obtain gene expression at up to sub-cellular resolution.
Disease Research
ST is particularly useful in disease research, such as cancer, where it may help to understand the tumor microenvironment and the interaction between cancer cells and other cells in the surrounding tissue.
Drug Discovery
ST can also aid in drug discovery by helping to identify how different regions of a tissue or tumor respond to a particular treatment.
Single-Cell Resolution
With advancements in technology, several versions of ST are now reaching single-cell and sub-cellular resolution, which provides even more detailed information about gene expression in individual cells within their spatial context.
Spatial Transcriptomics techniques
Slide sequencing
Thin sections of tissue are placed onto specially designed glass slides covered with a fixed array of DNA oligonucleotides. Each DNA oligo contains a unique barcode, and when the tissue is placed on the slide, the mRNA from the tissue is captured by spatially barcoded DNA oligos. The captured mRNA undergoes reverse transcription and is removed from the slide to generate DNA sequencing libraries. After sequencing, the origin of each mRNA molecule can be traced back to its original location in the tissue via the spatial barcode provided by the DNA capture oligos.
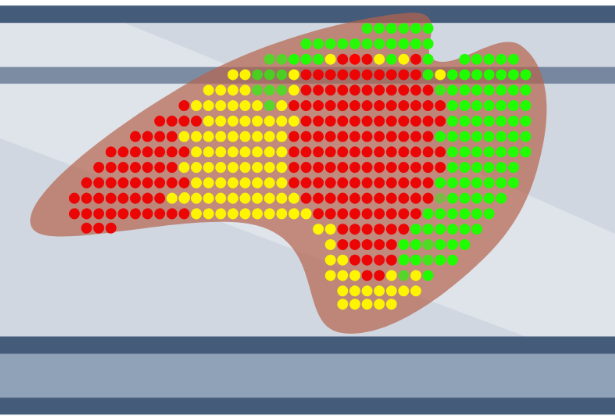
In situ
Slide-mounted tissue sections are exposed to pools of probes targeting mRNA, which then hybridize to mRNA within cells. Individual mRNA molecules are identified based on combinatorial barcodes generated through several rounds of labeling, imaging, and stripping. This approach can profile hundreds to thousands of mRNA molecules at single-cell and sub-cellular resolution.
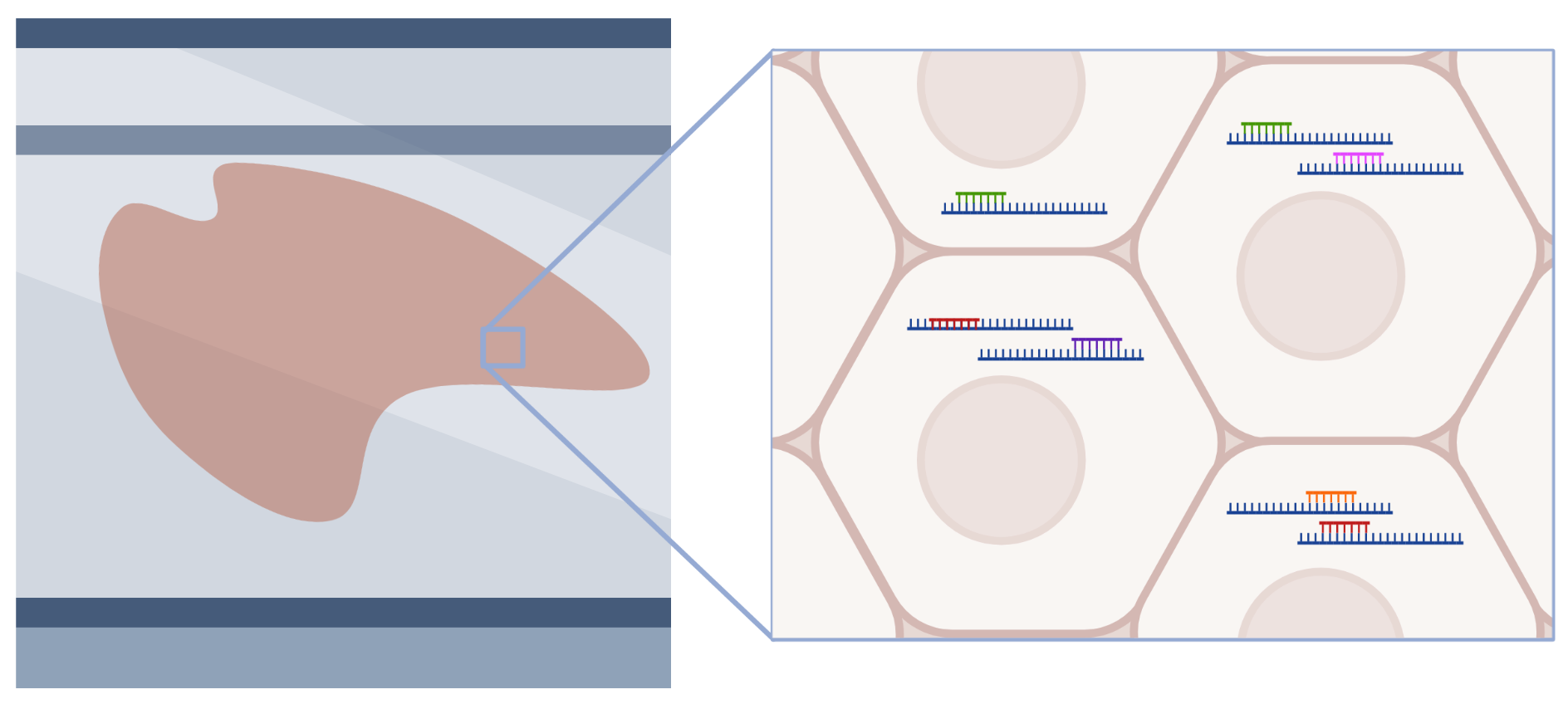
Spatial Transcriptomics - Seq workflow
- FFPE or fresh frozen tissue is transferred onto a specialized slide that contains a spatially barcoded DNA oligo array that serves to capture mRNA.
- Tissue is permeabilized, enabling mRNA to leak onto the DNA oligo array.
- mRNA undergoes reverse transcription and extension.
- cDNA is prepared into double-stranded DNA sequencing libraries.
- Libraries are read out using a DNA Next-Gen Sequencer.

- Data analysis enables mapping of mRNA to specific locations on the DNA array and tissue.
Spatial Transcriptomics - In situ workflow
- FFPE or fresh frozen tissue is mounted onto a microscope slide and pre-processed to prepare for staining with gene specific probes.
- Tissue is stained with transcript specific probes. The probes also contain reporter-probe binding sites used during imaging.
- mRNA is identified through a series of sequential probe binding-imaging-wash steps using a microscope. mRNA is identified by unique visual barcode detected during the rounds of imaging.
- Data analysis enables cell segmentation and cell classification.





